Ülkemizde 1 Haziran “Ulusal Fenilketonüri Günü” olarak kabul edilmiştir. Tedavi edilmediğinde sonuçları son derece ağır olan bu hastalık için toplumsal duyarlılık oluşturulmaya çalışılmaktadır. Anneler-Babalar ve adayları dikkat!
Sevgili anne babalar; tedavisi mümkün olan bu hastalığın tanısının konabilmesi için tüm yeni doğan bebeklerinize çok basit olan, ülkemizde tüm doğum yapılan hastanelerde ve Aile Hekimliklerinizde ücretsiz olarak yapılan Guithrie testini (halk arasında zekâ testi olarak söylenmektedir) yaptırmayı lütfen ihmal etmeyiniz.
Yenidoğan Tarama Programı, tüm dünyada gelişmiş ve gelişmekte olan ülkelerde halk sağlığı programları içerisinde çok önemli yeri olan koruyucu sağlık hizmetidir. Dünyada yenidoğanlarda ilk tarama programı 1962 yılında Amerika Birleşik Devletlerinde Massachusetts eyaletinde Fenilketonüri için başlatılmıştır. Dünyada yenidoğan döneminde en sık taraması yapılan hastalıklar arasında Fenilketonüri, Konjenital Hipotiroidi, Biotinidaz eksikliği, MSUD (Maple Syrup Urine Disease), Homosistinüri, Galaktozemi, Kongenital Adrenal Hiperplazi gibi metabolik ve endokrin hastalıklar yer almaktadır.

Türkiye’de Yenidoğan Taraması
Sağlık Bakanlığı’nın sorumluluğunda üniversitelerin desteğiyle 1987 Fenilketonüri Tarama Programı ile başlatılmıştır. Ülkemiz için yenidoğan taramasında milat olan Ulusal Yenidoğan Tarama Programı ise 25 Aralık 2006 tarihinde Sağlık Bakanlığı tarafından başlatılmıştır. 2008 Ekim ayı sonunda panele Biyotinidaz Eksikliği, 01 Ocak 2015 tarihinden itibaren de Kistik Fibrozis Hastalığı eklenmiştir.
Fenilketonüri kalıtsal metabolik bir hastalıktır. Hastalıkta bir protein yapıtaşı olan fenilalanin metabolize edilemez, kanda birikir ve geriye dönüşümsüz beyin hasarı yaratır. Erken tanımlanıp tedavi edilmediği takdirde kaçınılmaz son ağır zihinsel geriliktir. Ülkemiz hastalığın en sık izlendiği ülkelerdendir. Doğması beklenen bebek sayısı ile değerlendirildiğinde her yıl 200-250 yeni fenilketonüri vakasının topluma katılacağı hesaplanır. Çekinik genle taşınan bu hastalığın taşıyıcı sıklığı ülkemizde yüksektir. Her 100 kişiden dördünün bu hastalığı taşıyor olmasının yanı sıra, yüksek orandaki akraba evlilikleri (her 4 evlilikten 1’i) hastalığın ülkemizde sıklıkla izlenmesinin nedenidir.
Ağır zihinsel geriliği olan fenilketonürili bireylerde nörolojik sorunlar, bazı davranış bozuklukları, dermatit şeklindeki cilt lezyonları yanı sıra vakaların sadece % 60’ında anne babaya göre açık saç-göz-ten rengi ile karakterize görünüm vardır. Hastalığın tanısının ardından çocuklar uygun diyetle sağlıklı bir hayat sürebilmektedir.
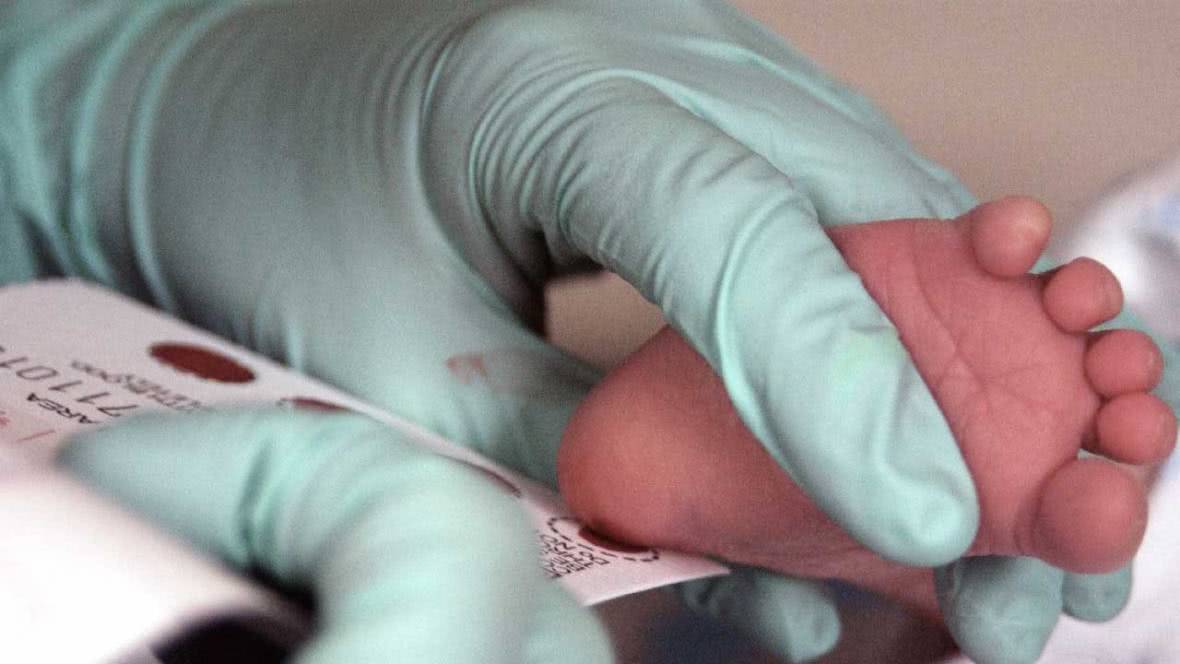
Yenidoğan Tarama Programı bebeklerin doğumlarından itibaren uygun şartlarda özel filtre kâğıtları ile topuk kanı örneklerinin alınarak Yenidoğan Tarama Laboratuvarlarına en kısa zamanda ulaştırması, bu bebeklerin tarama sonuçları internet ortamında açıklanır açıklanmaz sonuçları hastalık yönünden şüpheli çıkan bebekleri (Bilim Komisyonları tarafından hazırlanan akış şemaları uyarınca) ilgili kliniklere sevk etmek ve ilgili klinik tarafından verilen tanıları ve sonuçları takip etmek esasıyla yürütülmektedir.
2002 yılında taraması yapılan yenidoğan oranı %59,2 iken, tarama oranlarımız artık %95’lerin üstünde seyretmektedir. 2016 yılı tarama oranı % 97,5’dir.
Tanı alan fenilketonürileri çocukların özel beslenme gereksinimi olan mamaları Sosyal Güvenlik Kurumu tarafından karşılanmaktadır. Ayrıca ek beslenme ürünleri (makarna, bisküvi vb.) için ailelere ödeme de yapılmaktadır.
Ülkemizde 1 Haziran “Ulusal Fenilketonüri Günü” olarak kabul edilmiştir. Tedavi edilmediğinde sonuçları son derece ağır olan bu hastalık için toplumsal duyarlılık oluşturulmaya çalışılmaktadır.
Fenilketonüri, gıdalarla alınan ve esansiyel bir aminoasit olan fenilalenini tirozine çeviren fenilalanin hidroksilaz aktivitesinin yokluğu veya çok az olması sonucunda ortaya çıkan kalıtsal bir metabolik hastalıktır. Türkiye’de görülme sıklığı yaklaşık 1/3500’dür. Otozomal resesif (çekinik) olarak kalıtım yoluyla geçer.
Fenilalanin vücutta kullanılamadığı için fazla miktarlarda birikir ve başka yollarla fenilpirüvik asit veya feniletilamine dönüşür. Bu maddeler ve bunların metabolik ürünleri normal metabolizmayı bozarak beyin hasarına neden olurlar. Hastalığın ortaya çıkması için anne ve babanın her ikisinin de taşıyıcı ve/veya hasta olması gerekir (taşıyıcılarda hiçbir hastalık belirtisi bulunmaz). Bu nedenle akraba evliliklerinde risk daha yüksektir. Anne babanın her ikisinin de taşıyıcı olması durumunda, çocuklarının % 25 olasılıkla hasta doğma riski vardır.
Fenilketonüri hastalığı yenidoğan döneminde tanımlandığı takdirde tedavi edilebilen aksi halde ağır nörolojik ve gelişimsel bozukluklara ve zekâ geriliğine yol açan metabolik bir hastalıktır. Tanıda ve tedavide gecikme süresi ve tedaviye uyum hastalığın ağırlığını belirleyen en önemli unsurlardır. Hastalığın tanısı için yapılacaklar oldukça kolay, ucuz ve pratiktir. Yenidoğan döneminde kanda yükselmiş olan fenialalanin düzeyini tespit için bir tarama testi olan Guithrie’nin bakteriyel inhibisyon testi kullanılır. İnceleme için birkaç damla kan yeterlidir.
Fenilketonüri hastalığı olan bebeklerde kan fenilalenin düzeyi doğumdan hemen sonra beslenmeyi takiben ilk 4 saatte testi pozitif yapacak düzeylere yükselebilir. Ancak yalancı negatiflik olasılığını azaltmak için testin 3 günlük beslenme sonrası yapılması uygundur. Guithrie testinin pozitif olduğu bebeklerde ilk haftadan sonra plazma fenilalanin düzeyi kromatografik yöntemlerle saptanarak fenilketonüri hastalığının varlığı doğrulanmalıdır. Fenilketonüri, tanısı doğum öncesi konabilen bir hastalıktır. Bunun için amnios mayi hücreleri ya da korionik villusta DNA incelemeleri gerekir.
Türkiye’de fenilketonüri tarama testi, tüm sağlık kuruluşlarında ücretsiz olarak, yeni doğanlarda topuktan alınan bir damla kanın test kâğıdına emdirilerek sağlık bakanlığına gönderilmesi şeklinde uygulanmaktadır. Yalancı negatiflik riskini ortadan kaldırmak için tüm bebeklerde 1 - 2 haftalıkken Aile Hekimliklerinde testin tekrarlatılması önerilmektedir. Sonuçları şüpheli olanlar ve hastalığı olduğu saptananlar Aile Hekimlikleri aracılığıyla en kısa sürede bilgilendirilmekte, takip ve tedavisi planlanmak üzere üniversite hastanelerine yönlendirilmektedir.
Klinik bulgular: Fenilketonüri hastalığı bulunan bebekler doğumda klinik olarak tamamen normaldirler. En erken belirtilerden biri kusmalardır. Diğer erken belirti idrarın ve terin fare ve/ veya küf gibi kokmasıdır. Tedavi edilmeyen vakalarda 4 aylıktan itibaren sinir sistemi belirtileri ortaya çıkmaya başlar ve zamanla giderek artar. Çoğunda ağır zekâ ve nörolojik gelişim geriliği saptanır. Hiperaktivite, artmış kas tonusu, tremor ve % 25’inde konvülsif nöbetler görülmektedir. Hastaların çoğunun, melanin sentezindeki defekt nedeniyle, saç, cilt ve göz rengi açıktır.
Tedavi: Fenilketonüri hastalığında halen uygulanan tek tedavi yaşam boyu süren uygun gıdalarla beslenmedir. Beslenmeyi düzenlerken amaç; bir taraftan beyin hasarını önlemek veya minimuma indirmek, diğer taraftan diyetle hiperfenilaleninemiye yol açmayacak, ancak büyüme ve gelişme için yeterli fenilalanini sağlamaktır. Bunun için tanı konulur konulmaz; fenilalanini düşük, diğer aminoasitleri normal oranlarda içeren diyetler düzenlenir. Fenilketonürili bir bebeğin tedavisi; yaşam boyu sürer ve büyüme gelişme takibi, yaşına ve ihtiyaçlarına göre diyetinin ayarlanması, bu konuda yeterli deneyim ve donanıma sahip merkezlerin takibinde yapılmalıdır.





